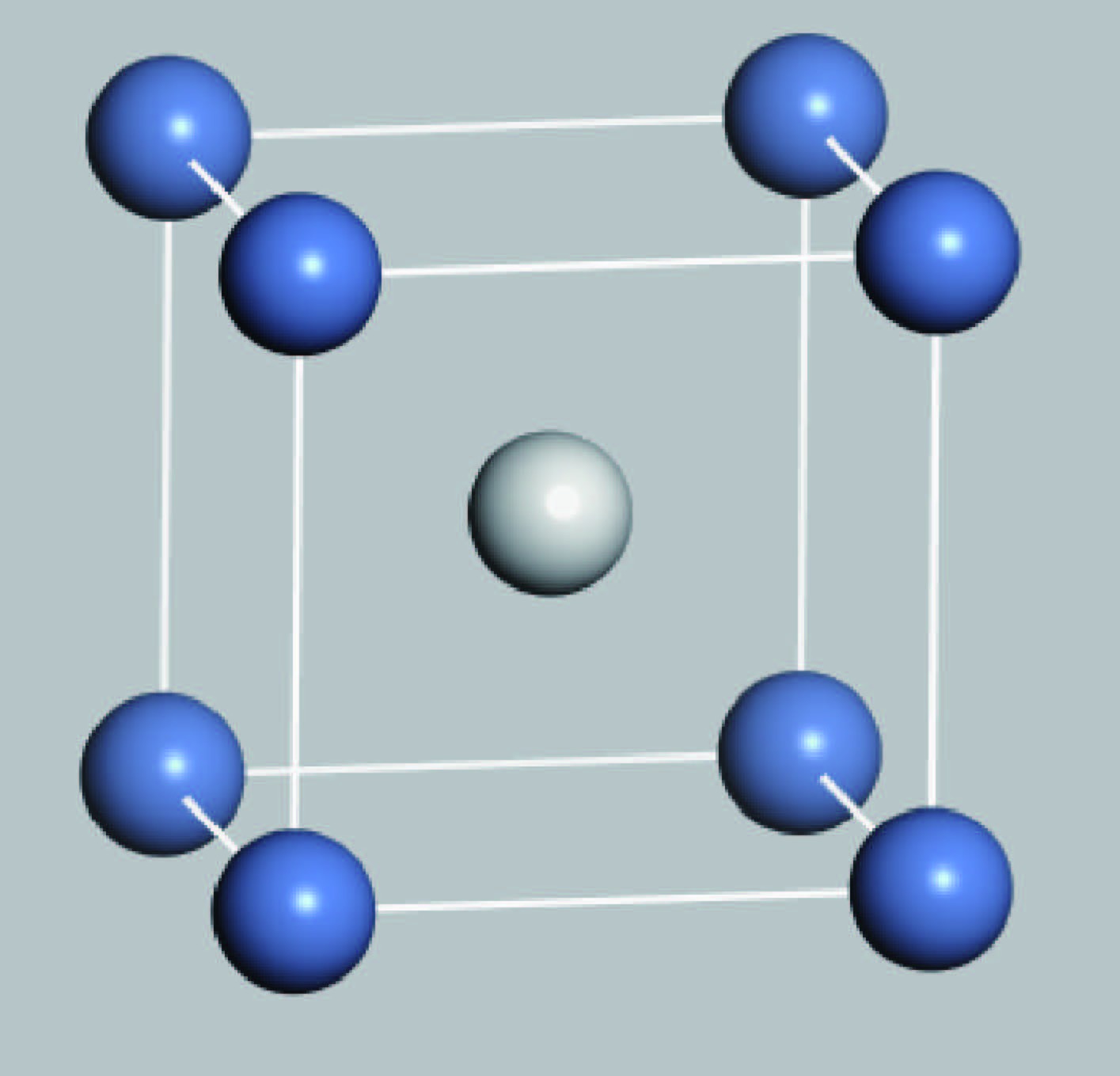
Fig. 1.
(Color online) Crystal structure of B2-NiSc (blue balls and white ball represent Ni atoms and Sc atom, respectively).
SEMICONDUCTOR PHYSICS
Zhipeng Yuan, Hongbao Cui and Xuefeng Guo
Corresponding author: Hongbao Cui,Email:cuihongbao@hpu.edu.cn
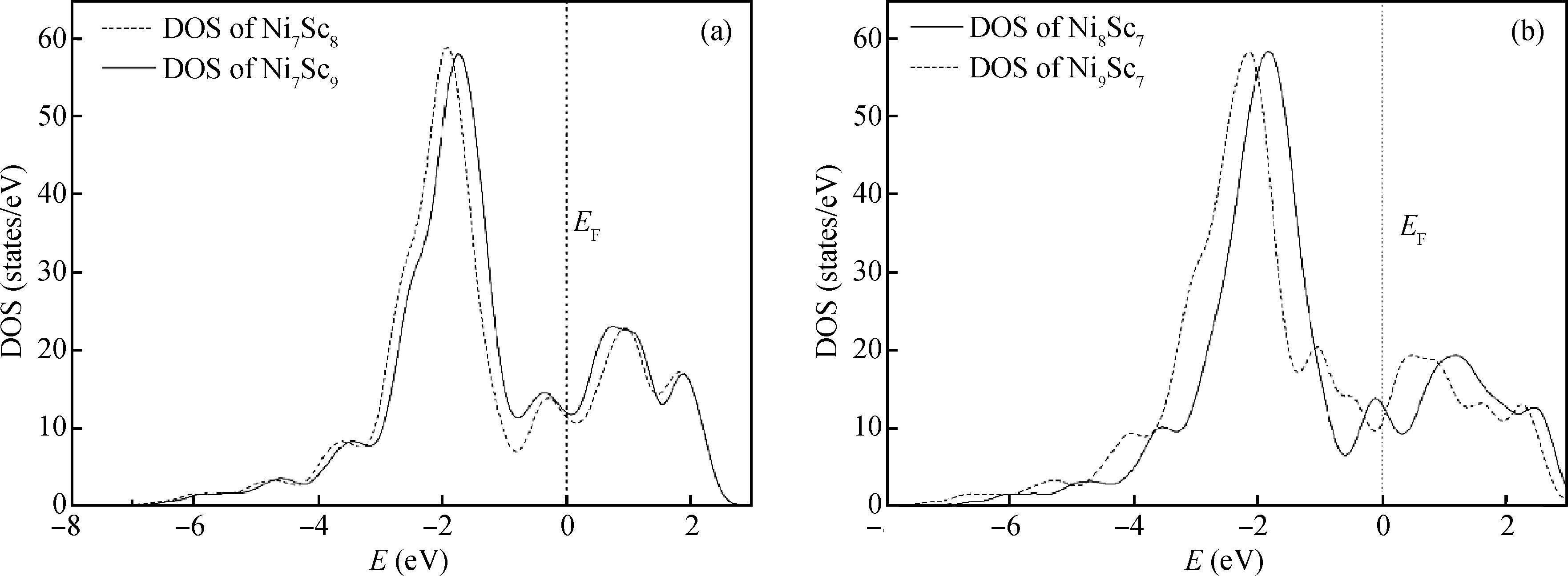
Abstract: Using the first-principles plane-wave pseudo-potential method based on density functional theory, the effect of vacancy and anti-position defect on the mechanical and thermal properties of B2-NiSc intermetallics were discussed in detail. Several parameters, such as the shear modulus, bulk modulus, modulus of elasticity, C11-C12, the Debye temperature and Poisson's ratio, have been calculated to evaluate the effect of vacancy and anti-position defect on the hardness, ductility and thermal properties of B2-NiSc intermetallics. The results show that VNi, ScNi, VSc and NiSc the four point defects all make the crystal hardness decrease and improve plasticity of B2-NiSc intermetallics. The entropy, enthalpy and free energy of VNi, ScNi, VSc and NiSc are monotonously changed as temperature changes. From the perspective of free energy, NiSc is the most stable, while ScNi is the most unstable. Debye temperature of NiSc intermetallics with four different point defects shows VNi, ScNi, VSc and NiSc the four point defects all reduce the stability of B2-NiSc intermetallics.
Key words: B2-NiSc intermetallics, vacancy, anti-position defect, plasticity, Debye temperature
| [1] |
Karl G, Alan R, Alexandra P, et al. A family of ductile intermetallic compounds. Nat Mater, 2003, 2(9): 587 doi: 10.1038/nmat958
|
| [2] |
Chen L, Peng P, Zhan J, et al. First-principles calculation on mechanical properties of B2-NiAl intermetallic compound with Fe addition. Rare Metal Mater Eng, 2010, 39(2): 229 http://en.cnki.com.cn/Article_en/CJFDTOTAL-COSE201002010.htm
|
| [3] |
Xie Z Y, Farkas D. Atomistic structure and lattice effects of vacancies in Ni-Al intermetallics. J Mater Res, 1994, 9(4): 875 doi: 10.1557/JMR.1994.0875
|
| [4] |
Fu C L, Ye Y, Yoo M H, et al. Equilibrium point defects in intermetallics with the B2 structure: NiAl and FeAl. Phys Rev B, 1993, 48(9): 6712 doi: 10.1103/PhysRevB.48.6712
|
| [5] |
Payne M C, Teter M P, Allan D C, et al. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev Mod Phys, 1992, 64(4): 1045 doi: 10.1103/RevModPhys.64.1045
|
| [6] |
Sun G P, Yan J L, Niu P J, et al. Electronic structure and optical property of p-type Zn-doped SnO2 with Sn vacancy. J Semicond, 2016, 37(2): 023005 doi: 10.1088/1674-4926/37/2/023005
|
| [7] |
Ma X G, Yan J, Liu N, et al. Effect of relaxation on the energetics and electronic structure of clean Ag3PO4(111) surface. J Semicond, 2016, 37(3): 033001 doi: 10.1088/1674-4926/37/3/033001
|
| [8] |
Liu Z Y, Lin D L, Huang B Y. Calculation on concentration of point defects in NiAl Intermetallics. J Shanghai Jiaotong Univ 1999, 33: 146
|
| [9] |
Thomas R M, Ann E M. Calculating the vacancy formation energy in metals: Pt, Pd, and Mo. Phys Rev B, 2002, 66(21): 214110 doi: 10.1103/PhysRevB.66.214110
|
| [10] |
Pan Y, Lin Y, Wang H, et al. Vacancy induced brittle-to-ductile transition of Nb5Si3 alloy from first-principles. Mater Des, 2015, 86: 259 http://www.docin.com/p-1322707578.html
|
| [11] |
Chen K Y, Zhao L R, John R, et al. Alloying effects on elastic properties of TiN-based nitride. J Phys D, 2003, 36(21): 2725 doi: 10.1088/0022-3727/36/21/021
|
| [12] |
Liu Q, Zhang R. Effect of 6.25 at% Al addition on structural stability of magnesium under high pressure: a first-principles study. J Alloys Compd, 2010, 508(2): 616 doi: 10.1016/j.jallcom.2010.08.142
|
| [13] |
Pugh S F. XCII. Relations between the modulus of elasticity and the plastic properties of polycrystalline pure metals. London Edinburgh Dublin Philos Mag J Sci, 2009, 45(367): 823 http://cn.bing.com/academic/profile?id=bbeea9069ccd86c3d749aa50e43fc021&encoded=0&v=paper_preview&mkt=zh-cn
|
| [14] |
Schiltz R J, Smith J F. Elastic constants of some MAl2 single crystals. J Appl Phys, 1974, 45(11): 4681 doi: 10.1063/1.1663118
|
| [15] |
Mattesini M, Ahuja R, Johansson B. Cubic Hf3N4 and Zr3N4: a class of hard materials. Phys Rev B, 2003, 68(18): 184108 doi: 10.1103/PhysRevB.68.184108
|
| [16] |
Anderson O L. A simplified method for calculating the Debye temperature from elastic constants. J Phys Chem Solids, 1963, 24(7): 909 doi: 10.1016/0022-3697(63)90067-2
|
| [17] |
Aydin S, Simsek M. First-principles calculations of MnB2, TcB2, and ReB2 within the ReB2-type structure. Phys Rev B, 2009, 80(13): 134107 doi: 10.1103/PhysRevB.80.134107
|
| [18] |
Hong S, Fu C L. Phase stability and elastic modulus of Cr2Nb by first-principles calculations. Intermetallics, 1999, 7(1): 5 doi: 10.1016/S0966-9795(98)00005-3
|
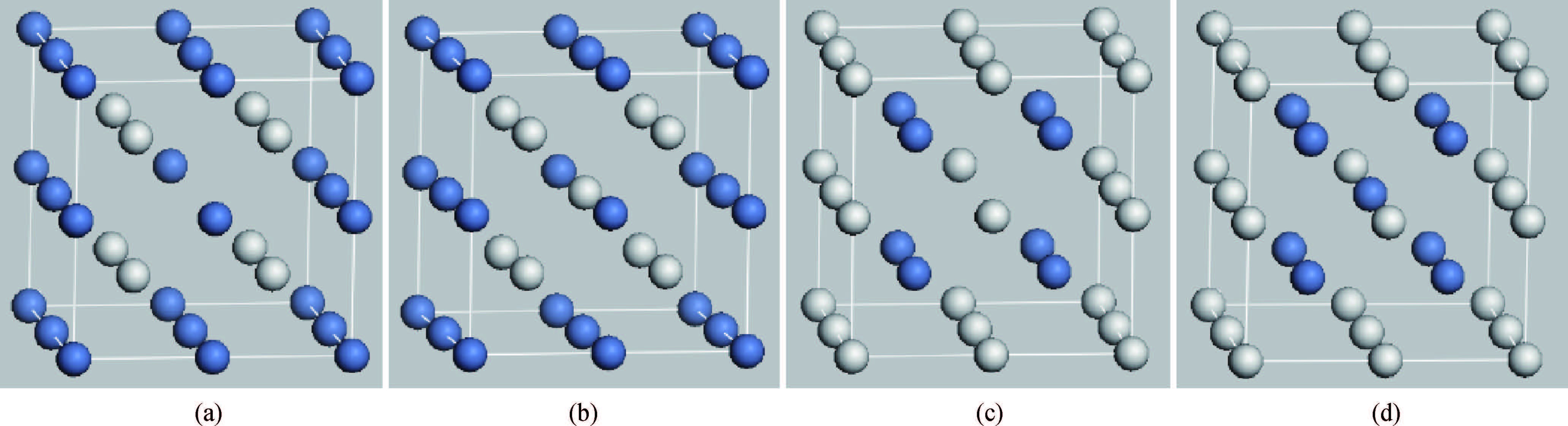

Table 1. The lattice constant of B2-NiSc without point defect and with four point defects.
| Point defect | This work (?) | Reference value (?) |
| No point defect | 3.171 | 3.171 |
| VNi | 3.164 | - |
| ScNi | 3.228 | - |
| VSc | 3.132 | - |
| NiSc | 3.123 | - |
 DownLoad: CSV
DownLoad: CSV
Table 2. The elastic constant of B2-NiSc without point defect and with four point defects.
| Elastic constant | No point defect | VNi | ScNi | VSc | NiSc |
| $C_{11}$ | 187.76 | 123.30 | 144.80 | 108.79 | 130.79 |
| $C_{12}$ | 70.03 | 78.68 | 74.16 | 70.02 | 108.15 |
| $C_{44}$ | 51.40 | 36.07 | 45.02 | 42.83 | 53.20 |
DownLoad: CSV
Table 3. The shear modulus, bulk modulus and modulus of elasticity of B2-NiSc without point defect and with four point defects, respectively.
| Parameter | No point defect | VNi | ScNi | VSc | NiSc |
| G (GPa) | 54.27 | 29.75 | 40.85 | 31.16 | 28.95 |
| B (GPa) | 109.28 | 93.56 | 97.71 | 82.94 | 115.69 |
| E (GPa) | 139.69 | 80.70 | 107.56 | 83.08 | 80.16 |
DownLoad: CSV
Table 4. The $C_{11}-C_{12}$, G/B ratio and Poisson's ratio of B2-NiSc without point defect and with four point defects.
| Parameter | No point defect | VNi | ScNi | VSc | NiSc |
| $C_{11}-C_{12}$ (GPa) | 80.62 | 44.62 | 43.90 | 38.77 | 22.64 |
| $G/B $ | 0.50 | 0.32 | 0.34 | 0.38 | 0.25 |
| $nu $ | 0.29 | 0.36 | 0.35 | 0.33 | 0.39 |
DownLoad: CSV
| [1] |
Karl G, Alan R, Alexandra P, et al. A family of ductile intermetallic compounds. Nat Mater, 2003, 2(9): 587 doi: 10.1038/nmat958
|
| [2] |
Chen L, Peng P, Zhan J, et al. First-principles calculation on mechanical properties of B2-NiAl intermetallic compound with Fe addition. Rare Metal Mater Eng, 2010, 39(2): 229 http://en.cnki.com.cn/Article_en/CJFDTOTAL-COSE201002010.htm
|
| [3] |
Xie Z Y, Farkas D. Atomistic structure and lattice effects of vacancies in Ni-Al intermetallics. J Mater Res, 1994, 9(4): 875 doi: 10.1557/JMR.1994.0875
|
| [4] |
Fu C L, Ye Y, Yoo M H, et al. Equilibrium point defects in intermetallics with the B2 structure: NiAl and FeAl. Phys Rev B, 1993, 48(9): 6712 doi: 10.1103/PhysRevB.48.6712
|
| [5] |
Payne M C, Teter M P, Allan D C, et al. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev Mod Phys, 1992, 64(4): 1045 doi: 10.1103/RevModPhys.64.1045
|
| [6] |
Sun G P, Yan J L, Niu P J, et al. Electronic structure and optical property of p-type Zn-doped SnO2 with Sn vacancy. J Semicond, 2016, 37(2): 023005 doi: 10.1088/1674-4926/37/2/023005
|
| [7] |
Ma X G, Yan J, Liu N, et al. Effect of relaxation on the energetics and electronic structure of clean Ag3PO4(111) surface. J Semicond, 2016, 37(3): 033001 doi: 10.1088/1674-4926/37/3/033001
|
| [8] |
Liu Z Y, Lin D L, Huang B Y. Calculation on concentration of point defects in NiAl Intermetallics. J Shanghai Jiaotong Univ 1999, 33: 146
|
| [9] |
Thomas R M, Ann E M. Calculating the vacancy formation energy in metals: Pt, Pd, and Mo. Phys Rev B, 2002, 66(21): 214110 doi: 10.1103/PhysRevB.66.214110
|
| [10] |
Pan Y, Lin Y, Wang H, et al. Vacancy induced brittle-to-ductile transition of Nb5Si3 alloy from first-principles. Mater Des, 2015, 86: 259 http://www.docin.com/p-1322707578.html
|
| [11] |
Chen K Y, Zhao L R, John R, et al. Alloying effects on elastic properties of TiN-based nitride. J Phys D, 2003, 36(21): 2725 doi: 10.1088/0022-3727/36/21/021
|
| [12] |
Liu Q, Zhang R. Effect of 6.25 at% Al addition on structural stability of magnesium under high pressure: a first-principles study. J Alloys Compd, 2010, 508(2): 616 doi: 10.1016/j.jallcom.2010.08.142
|
| [13] |
Pugh S F. XCII. Relations between the modulus of elasticity and the plastic properties of polycrystalline pure metals. London Edinburgh Dublin Philos Mag J Sci, 2009, 45(367): 823 http://cn.bing.com/academic/profile?id=bbeea9069ccd86c3d749aa50e43fc021&encoded=0&v=paper_preview&mkt=zh-cn
|
| [14] |
Schiltz R J, Smith J F. Elastic constants of some MAl2 single crystals. J Appl Phys, 1974, 45(11): 4681 doi: 10.1063/1.1663118
|
| [15] |
Mattesini M, Ahuja R, Johansson B. Cubic Hf3N4 and Zr3N4: a class of hard materials. Phys Rev B, 2003, 68(18): 184108 doi: 10.1103/PhysRevB.68.184108
|
| [16] |
Anderson O L. A simplified method for calculating the Debye temperature from elastic constants. J Phys Chem Solids, 1963, 24(7): 909 doi: 10.1016/0022-3697(63)90067-2
|
| [17] |
Aydin S, Simsek M. First-principles calculations of MnB2, TcB2, and ReB2 within the ReB2-type structure. Phys Rev B, 2009, 80(13): 134107 doi: 10.1103/PhysRevB.80.134107
|
| [18] |
Hong S, Fu C L. Phase stability and elastic modulus of Cr2Nb by first-principles calculations. Intermetallics, 1999, 7(1): 5 doi: 10.1016/S0966-9795(98)00005-3
|









Article views: 3862 Times PDF downloads: 28 Times Cited by: 0 Times
Received: 09 May 2016 Revised: 30 June 2016 Online: Published: 01 January 2017
| Citation: |
Zhipeng Yuan, Hongbao Cui, Xuefeng Guo. First-principle calculation on mechanical and thermal properties of B2-NiSc with point defects[J]. Journal of Semiconductors, 2017, 38(1): 012001. doi: 10.1088/1674-4926/38/1/012001
****
Z P Yuan, H B Cui, X F Guo. First-principle calculation on mechanical and thermal properties of B2-NiSc with point defects[J]. J. Semicond., 2017, 38(1): 012001. doi: 10.1088/1674-4926/38/1/012001.

|
Project supported by the National Natural Science Foundation of China Nos. 51301063, 51571086
and the Talent Introduction Foundation of Henan Polytechnic University No. Y-2009
Project supported by the National Natural Science Foundation of China (Nos. 51301063, 51571086) and the Talent Introduction Foundation of Henan Polytechnic University (No. Y-2009).
| [1] |
Karl G, Alan R, Alexandra P, et al. A family of ductile intermetallic compounds. Nat Mater, 2003, 2(9): 587 doi: 10.1038/nmat958
|
| [2] |
Chen L, Peng P, Zhan J, et al. First-principles calculation on mechanical properties of B2-NiAl intermetallic compound with Fe addition. Rare Metal Mater Eng, 2010, 39(2): 229 http://en.cnki.com.cn/Article_en/CJFDTOTAL-COSE201002010.htm
|
| [3] |
Xie Z Y, Farkas D. Atomistic structure and lattice effects of vacancies in Ni-Al intermetallics. J Mater Res, 1994, 9(4): 875 doi: 10.1557/JMR.1994.0875
|
| [4] |
Fu C L, Ye Y, Yoo M H, et al. Equilibrium point defects in intermetallics with the B2 structure: NiAl and FeAl. Phys Rev B, 1993, 48(9): 6712 doi: 10.1103/PhysRevB.48.6712
|
| [5] |
Payne M C, Teter M P, Allan D C, et al. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev Mod Phys, 1992, 64(4): 1045 doi: 10.1103/RevModPhys.64.1045
|
| [6] |
Sun G P, Yan J L, Niu P J, et al. Electronic structure and optical property of p-type Zn-doped SnO2 with Sn vacancy. J Semicond, 2016, 37(2): 023005 doi: 10.1088/1674-4926/37/2/023005
|
| [7] |
Ma X G, Yan J, Liu N, et al. Effect of relaxation on the energetics and electronic structure of clean Ag3PO4(111) surface. J Semicond, 2016, 37(3): 033001 doi: 10.1088/1674-4926/37/3/033001
|
| [8] |
Liu Z Y, Lin D L, Huang B Y. Calculation on concentration of point defects in NiAl Intermetallics. J Shanghai Jiaotong Univ 1999, 33: 146
|
| [9] |
Thomas R M, Ann E M. Calculating the vacancy formation energy in metals: Pt, Pd, and Mo. Phys Rev B, 2002, 66(21): 214110 doi: 10.1103/PhysRevB.66.214110
|
| [10] |
Pan Y, Lin Y, Wang H, et al. Vacancy induced brittle-to-ductile transition of Nb5Si3 alloy from first-principles. Mater Des, 2015, 86: 259 http://www.docin.com/p-1322707578.html
|
| [11] |
Chen K Y, Zhao L R, John R, et al. Alloying effects on elastic properties of TiN-based nitride. J Phys D, 2003, 36(21): 2725 doi: 10.1088/0022-3727/36/21/021
|
| [12] |
Liu Q, Zhang R. Effect of 6.25 at% Al addition on structural stability of magnesium under high pressure: a first-principles study. J Alloys Compd, 2010, 508(2): 616 doi: 10.1016/j.jallcom.2010.08.142
|
| [13] |
Pugh S F. XCII. Relations between the modulus of elasticity and the plastic properties of polycrystalline pure metals. London Edinburgh Dublin Philos Mag J Sci, 2009, 45(367): 823 http://cn.bing.com/academic/profile?id=bbeea9069ccd86c3d749aa50e43fc021&encoded=0&v=paper_preview&mkt=zh-cn
|
| [14] |
Schiltz R J, Smith J F. Elastic constants of some MAl2 single crystals. J Appl Phys, 1974, 45(11): 4681 doi: 10.1063/1.1663118
|
| [15] |
Mattesini M, Ahuja R, Johansson B. Cubic Hf3N4 and Zr3N4: a class of hard materials. Phys Rev B, 2003, 68(18): 184108 doi: 10.1103/PhysRevB.68.184108
|
| [16] |
Anderson O L. A simplified method for calculating the Debye temperature from elastic constants. J Phys Chem Solids, 1963, 24(7): 909 doi: 10.1016/0022-3697(63)90067-2
|
| [17] |
Aydin S, Simsek M. First-principles calculations of MnB2, TcB2, and ReB2 within the ReB2-type structure. Phys Rev B, 2009, 80(13): 134107 doi: 10.1103/PhysRevB.80.134107
|
| [18] |
Hong S, Fu C L. Phase stability and elastic modulus of Cr2Nb by first-principles calculations. Intermetallics, 1999, 7(1): 5 doi: 10.1016/S0966-9795(98)00005-3
|
 WeChat ID
WeChat ID
Journal of Semiconductors © 2017 All Rights Reserved 京ICP備05085259號-2